【健康科普】为爱呐“罕” ——带你认识罕见病,进行性肌营养不良
时间:2024年01月08日 点击次数:218
王民杰
什么是罕见病?
“罕见病”是一大类发病率很低、很少见的疾病,世卫组织定义为患病人数在0.65‰-1‰的疾病,目前已经确认的约7000种。
在中国,罕见病指患病率低于1/50万或新生儿发病率低于1/10000,又称“孤儿病”。目前治疗罕见病的药物极其有限,只有5%有药可治,且往往因其治疗费用昂贵,为患者及家属带来了沉重的精神与经济负担。
近期,我院儿科接诊几例以“肝脏转氨酶增高”“心肌酶增高”就诊于多家医院的患儿,最终确诊为“进行性肌营养不良”。在此,让我们一起来认识这种罕见疾病吧。
什么是进行性肌营养不良?
进行性肌营养不良是一组早期发病的遗传性疾病,常表现为以近端受累为主的骨骼肌进行性无力、肌萎缩、假性肌肥大,可最终完全丧失运动功能。根据遗传方式、起病年龄、受累肌群、病程进展与预后等因素,分为六种主要亚型,其中以杜氏型(假性肥大型)最多见。
进行性肌营养不良有哪些临床表现?
Duchenne/Becker型肌营养不良(DMD/BMD)是最常见的两种类型,为X连锁隐性遗传,是男性中常见的遗传性疾病,我国的发病率约为1/3853。
Duchenne型肌营养不良常在儿童期起病,主要表现为运动发育轻度迟滞,骨骼肌进行性无力萎缩,影响肢体运动功能,逐渐出现步态异常、上肢活动受限,患儿常在10岁左右丧失行走能力。此后出现脊柱侧弯、关节挛缩、呼吸肌无力、扩张性心肌病,20岁左右因呼吸衰竭、心功能衰竭死亡。查体可见双腓肠肌假肥大,同时可有双前臂及舌肌假肥大,Gower’s征阳性,腰椎前凸等。
Becker型肌营养不良为同一疾病的相对良性表型,通常青年或成年起病,部分患者不影响生存期。假肥大体征明显,部分患者在肢体无力尚轻时,先出现明显的扩张性心肌病。
其他类型还有面肩肱型肌营养不良1型、面肩肱型肌营养不良2型、肢带型肌营养不良、眼咽型肌营养不良、Emery-Dreifuss型肌营养不良、先天性肌营养不良、远端型肌营养不良等。
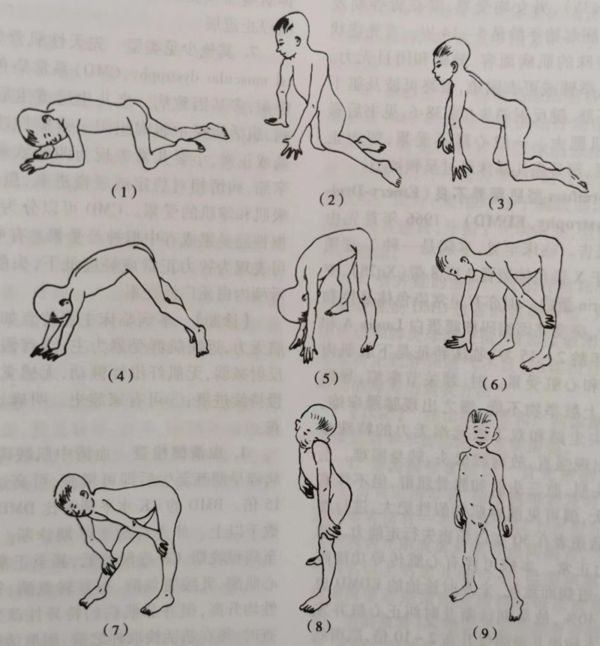
进行性肌营养不良小儿自卧位至起立位的动作步骤
进行性肌营养不良如何诊断?
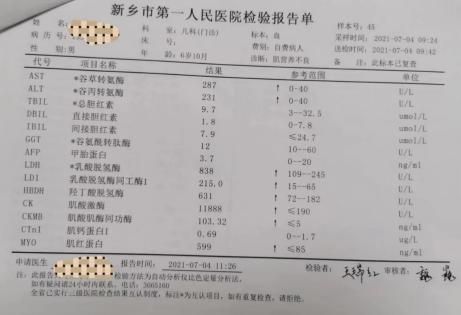
对疑诊进行性肌营养不良患者一般需通过完善以下辅助检查以帮助明确诊断。血清学检测可发现肌酸激酶、乳酸脱氢酶、肌红蛋白等明显升高,可达正常上限的20~200倍。
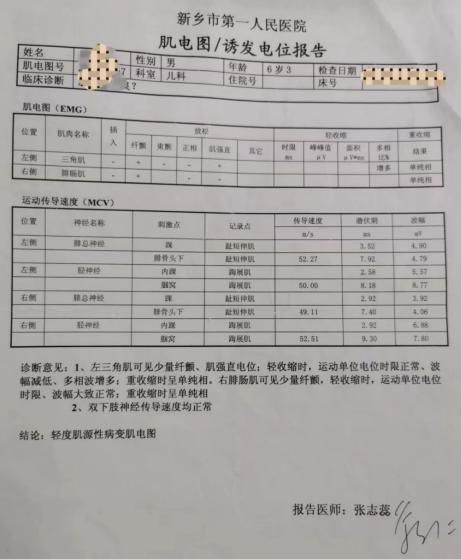
肌电图检查可见纤颤波、正锐波、多相波及强直样放电等典型的肌源性受损的表现。肌肉MR可发现肌肉组织中存在炎性水肿和(或)脂肪替代,同时明确骨骼肌受累的范围。
 肌肉活检可发现肌肉的坏死再生、间质脂肪和纤维结缔组织增生。基因检测对DMD/BMD诊断具有重要价值。基因检测有多种不同方法,疑诊DMD/BMD,一般先用多重连接探针扩增技术(MLPA)检测DMD基因大片段缺失和重复,如果未发现此类拷贝数异常,再用高通量测序技术(NGS)检测微小突变。
肌肉活检可发现肌肉的坏死再生、间质脂肪和纤维结缔组织增生。基因检测对DMD/BMD诊断具有重要价值。基因检测有多种不同方法,疑诊DMD/BMD,一般先用多重连接探针扩增技术(MLPA)检测DMD基因大片段缺失和重复,如果未发现此类拷贝数异常,再用高通量测序技术(NGS)检测微小突变。

进行性肌营养不良的治疗?
进行性肌营养不良症迄今无特异性治疗,只能对症治疗及支持治疗,如增加营养、适当锻炼等。
物理疗法和矫形治疗可预防及改善脊柱畸形和关节挛缩,尤其是早期进行踝关节挛缩的矫正,对维持行走功能很重要。应鼓励患者尽可能从事日常活动,避免长期卧床。
药物可选用ATP、肌苷、维生素E、肌生注射液和补中益气的通塞脉片等。
基因治疗(外显子跳跃、微小基因替代)及干细胞移植治疗有望成为有效的治疗方法。由于目前尚无有效的治疗方法,因此检出携带者、进行产前诊断就尤为重要。
什么是罕见病?
“罕见病”是一大类发病率很低、很少见的疾病,世卫组织定义为患病人数在0.65‰-1‰的疾病,目前已经确认的约7000种。
在中国,罕见病指患病率低于1/50万或新生儿发病率低于1/10000,又称“孤儿病”。目前治疗罕见病的药物极其有限,只有5%有药可治,且往往因其治疗费用昂贵,为患者及家属带来了沉重的精神与经济负担。
近期,我院儿科接诊几例以“肝脏转氨酶增高”“心肌酶增高”就诊于多家医院的患儿,最终确诊为“进行性肌营养不良”。在此,让我们一起来认识这种罕见疾病吧。
什么是进行性肌营养不良?
进行性肌营养不良是一组早期发病的遗传性疾病,常表现为以近端受累为主的骨骼肌进行性无力、肌萎缩、假性肌肥大,可最终完全丧失运动功能。根据遗传方式、起病年龄、受累肌群、病程进展与预后等因素,分为六种主要亚型,其中以杜氏型(假性肥大型)最多见。
进行性肌营养不良有哪些临床表现?
Duchenne/Becker型肌营养不良(DMD/BMD)是最常见的两种类型,为X连锁隐性遗传,是男性中常见的遗传性疾病,我国的发病率约为1/3853。
Duchenne型肌营养不良常在儿童期起病,主要表现为运动发育轻度迟滞,骨骼肌进行性无力萎缩,影响肢体运动功能,逐渐出现步态异常、上肢活动受限,患儿常在10岁左右丧失行走能力。此后出现脊柱侧弯、关节挛缩、呼吸肌无力、扩张性心肌病,20岁左右因呼吸衰竭、心功能衰竭死亡。查体可见双腓肠肌假肥大,同时可有双前臂及舌肌假肥大,Gower’s征阳性,腰椎前凸等。
Becker型肌营养不良为同一疾病的相对良性表型,通常青年或成年起病,部分患者不影响生存期。假肥大体征明显,部分患者在肢体无力尚轻时,先出现明显的扩张性心肌病。
其他类型还有面肩肱型肌营养不良1型、面肩肱型肌营养不良2型、肢带型肌营养不良、眼咽型肌营养不良、Emery-Dreifuss型肌营养不良、先天性肌营养不良、远端型肌营养不良等。
进行性肌营养不良小儿自卧位至起立位的动作步骤
进行性肌营养不良如何诊断?
对疑诊进行性肌营养不良患者一般需通过完善以下辅助检查以帮助明确诊断。血清学检测可发现肌酸激酶、乳酸脱氢酶、肌红蛋白等明显升高,可达正常上限的20~200倍。
肌电图检查可见纤颤波、正锐波、多相波及强直样放电等典型的肌源性受损的表现。肌肉MR可发现肌肉组织中存在炎性水肿和(或)脂肪替代,同时明确骨骼肌受累的范围。
进行性肌营养不良的治疗?
进行性肌营养不良症迄今无特异性治疗,只能对症治疗及支持治疗,如增加营养、适当锻炼等。
物理疗法和矫形治疗可预防及改善脊柱畸形和关节挛缩,尤其是早期进行踝关节挛缩的矫正,对维持行走功能很重要。应鼓励患者尽可能从事日常活动,避免长期卧床。
药物可选用ATP、肌苷、维生素E、肌生注射液和补中益气的通塞脉片等。
基因治疗(外显子跳跃、微小基因替代)及干细胞移植治疗有望成为有效的治疗方法。由于目前尚无有效的治疗方法,因此检出携带者、进行产前诊断就尤为重要。